Researchers have developed a novel approach to accurately simulate the complex energy transfer processes between molecules and metal surfaces, paving the way for advancements in fields like catalysis, materials science, and nanotechnology.
Bridging the Gap in Modeling Nonadiabatic Dynamics
It has been proven by countless experimental observations that the non-adiabatical excitation energy transfer is a ubiquitous process in interfacial systems. Such energy transfer processes are relevant to many fundamental phenomena, including chemical adsorption, electrochemistry and plasmonic catalysis.
However, the interaction of molecules with metal surfaces is highly intricate and complex, where vibrational, rotational and translational degrees of freedom in the molecule couple to surface phonons and electrons. Ultimately it presents a variety of interesting results however traditional modelsthmus far through electronic friction leave something to be desired given the seemingly complex energy transfer dynamics witnessed in experimental studies.
A Multifaceted Approach to Simulation
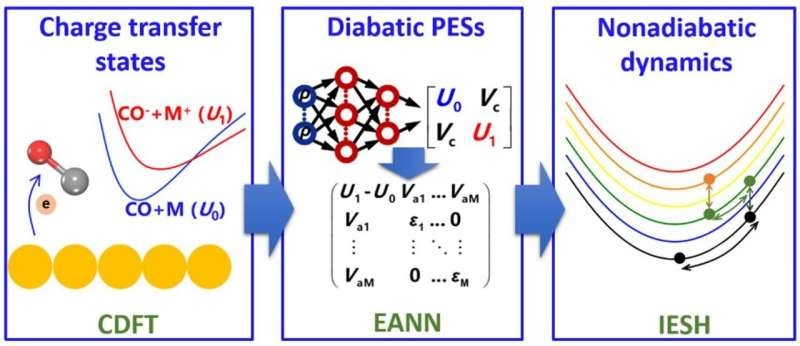
Now, for the first time, a research team led by Prof. Jiang Bin from University of Science and Technology of China of Chinese Academy of Sciences develops an integrated simulation method to handle this grand challenging scientific issue. This technique groups the following important steps together:
1. Charge-transfer states of CO molecule, with different binding geometries at the metal surfaces, are calculated by the CDFT.
2. Learning the CDFT energies and generating high-dimensional diabatic potential energy surfaces (PESs) with an embedded atom neural network (EANN).
3. Simulations of the energy transfer process using the independent electron surface hopping (IESH) in different regimes.
Conclusion
This new technique for simulation is said to be a game-changing tool for improving the understanding of energy transfer in molecule-surface systems. This will allow for the application of our approach to a broad class of nonadiabatic surface phenomena and ultimately has the potential to revolutionize fields ranging from catalysis, materials science, and nanotechnology. As the researchers further perfect and broaden this framework, we will be able to see much more deeply into how molecules dance with metal surfaces — and that knowledge could guide transformative advances in applications dependent on these fundamental interactions.