Researchers have shed new light on the genetic mutations behind Glanzmann thrombasthenia, a rare inherited bleeding disorder. By using advanced computational techniques, they’ve discovered how specific mutations in the integrin αIIbβ3 protein can disrupt its structure and function, leading to the severe symptoms experienced by some patients. This groundbreaking study provides valuable insights that could pave the way for improved diagnostics and personalized treatments for this debilitating condition.

Unraveling the Genetic Mysteries of Glanzmann Thrombasthenia
Glanzmann thrombasthenia is a rare genetic disorder that affects the ability of blood platelets to clot properly, leading to excessive bleeding and bruising. This condition is caused by mutations in the genes that encode the integrin αIIbβ3 protein, which plays a crucial role in platelet aggregation during the clotting process.
Computational Insights into Protein Structure and Function
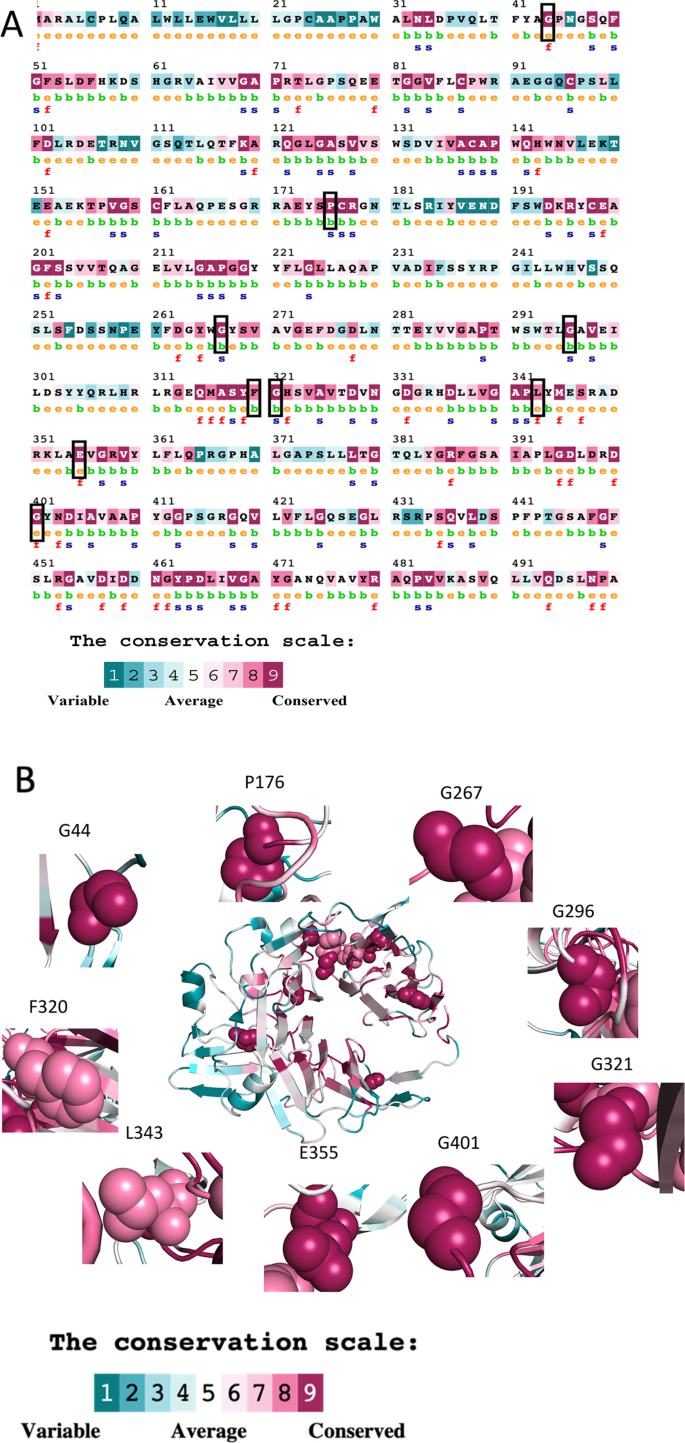
In a recent study, researchers set out to investigate the impact of various mutations in the β-propeller domain of the αIIb subunit of the integrin αIIbβ3 protein. By combining data from multiple genetic databases and employing advanced computational techniques, such as molecular dynamics simulations, the researchers gained valuable insights into how these mutations can disrupt the structure and function of the protein.
Uncovering the Impact of Specific Mutations
The researchers focused on two particular mutations, E355K and G401C, which had been previously reported in patients with Glanzmann thrombasthenia. Their findings revealed that these mutations can have significant consequences on the stability and binding properties of the integrin αIIbβ3 complex, ultimately affecting its ability to interact with its primary ligand, fibrinogen.
Linking Genotype to Phenotype
The E355K mutation, found in a Swiss patient with a severe form of the disease, was shown to cause substantial structural changes and reduced binding affinity for fibrinogen. In contrast, the G401C mutation, reported in a French patient with a milder clinical presentation, had a comparatively smaller impact on the protein’s stability and ligand binding.
Implications for Improved Diagnosis and Targeted Therapy
This comprehensive in silico analysis not only highlights the critical role of the β-propeller domain in the structure and function of the integrin αIIbβ3 but also provides a direct link between genotype and phenotype in Glanzmann thrombasthenia. These findings could pave the way for more accurate genetic diagnosis and the development of personalized therapeutic strategies for patients with this rare bleeding disorder.
By unraveling the molecular mechanisms underlying Glanzmann thrombasthenia, this study represents a significant step forward in our understanding of the genetic and structural factors that contribute to this debilitating condition. The insights gained from this research could have far-reaching implications for improving the lives of individuals affected by this rare but challenging disorder.
Author credit: This article is based on research by Finola Priyadharshini Chandrasekaran, Everette Jacob Remington Nelson.
For More Related Articles Click Here