Ciliopathies are a group of complex disorders that arise from abnormalities in the primary cilium, a crucial cellular structure involved in various signaling pathways. While significant research has focused on the role of the primary cilium in conditions like Meckel-Gruber and Joubert syndromes, the underlying causes of some ciliopathy-related disorders, such as liver malformations, have remained elusive. Primary cilium and ciliopathies play a crucial role in this new discovery.
In a groundbreaking study, researchers have uncovered a previously unknown function of the B9D2 protein, a key component of the primary cilium’s transition zone. Surprisingly, they found that B9D2 plays a crucial role in the maturation and maintenance of tight junctions, which are essential for the formation and integrity of epithelial barriers, including the bile ducts. This discovery offers new insights into the mechanisms underlying the development of biliary dysgenesis, a condition commonly observed in liver-related ciliopathies.

Main Content:
Unraveling the Complexities of Ciliopathies
Ciliopathies are a diverse group of disorders that arise from abnormalities in the development or function of the primary cilium, a hair-like structure found on the surface of many cells. This “cellular antenna” plays a crucial role in transmitting external signals, such as biochemical, osmotic, and mechanical cues, which are essential for proper organ development and function.
Meckel-Gruber syndrome and Joubert syndrome are two well-known ciliopathies that are characterized by a range of developmental abnormalities, including central nervous system issues, polycystic kidneys, liver malformations, and polydactyly (extra fingers or toes). While significant progress has been made in understanding the role of the primary cilium in these conditions, the underlying causes of some ciliopathy-related disorders, particularly those affecting the liver, have remained elusive.
Uncovering the Extraciliary Functions of B9D2
In this groundbreaking study, researchers focused on the B9D2 protein, a key component of the primary cilium’s transition zone. The transition zone is a specialized region at the base of the cilium that acts as a gatekeeper, regulating the entry and exit of proteins essential for ciliary function.
Surprisingly, the researchers discovered that B9D2 plays a crucial role in the maturation and maintenance of tight junctions, which are specialized cell-cell adhesions that form a critical barrier in epithelial tissues, such as the bile ducts. This finding suggests that B9D2 has important extraciliary functions beyond its well-known role in the primary cilium.

The Crucial Role of B9D2 in Tight Junction Integrity
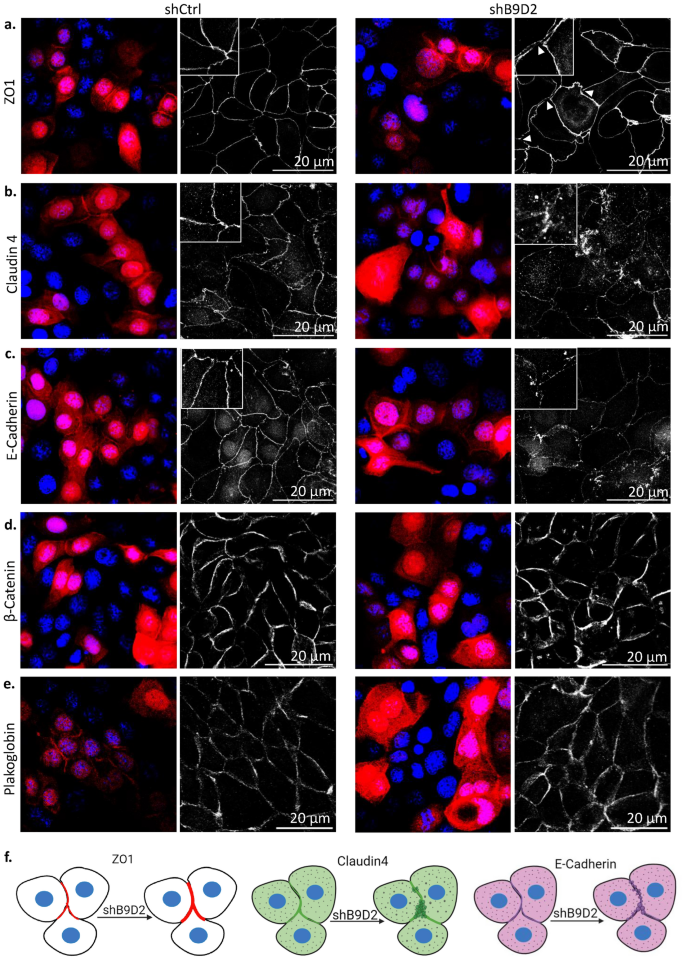
Through a series of experiments, the researchers demonstrated that B9D2 localizes to the tight junctions in polarized epithelial cells, even before the formation of the primary cilium. They found that B9D2 is essential for the proper localization and organization of key tight junction proteins, such as ZO1, Claudin 4, and E-cadherin.
When B9D2 expression was knocked down, the researchers observed significant disruptions in the structure and function of the tight junctions. The ZO1 protein, which normally forms a thin, continuous belt around the cells, appeared thicker and more discontinuous. Additionally, the transmembrane proteins Claudin 4 and E-cadherin were mislocalized, no longer concentrated at the cell-cell junctions.
These findings suggest that B9D2 plays a critical role in maintaining the integrity and organization of tight junctions, which are essential for the formation of a functional epithelial barrier.

Implications for Bile Duct Development and Ciliopathies
The researchers’ discovery of B9D2’s extraciliary functions in tight junction maintenance has important implications for understanding the development of bile ducts and the pathogenesis of liver-related ciliopathies.
During embryonic development, the bile ducts form from a structure called the ductal plate, which undergoes remodeling to create an interconnected network of bile ducts. Patients with Meckel-Gruber or Joubert syndrome often exhibit abnormalities in this process, leading to dilated and mispositioned bile ducts, as well as the presence of cysts and liver fibrosis.
The researchers hypothesize that the disruption of tight junction integrity caused by B9D2 deficiency may be a key factor in the development of these biliary malformations observed in liver-related ciliopathies. By impairing the maturation and expansion of the bile duct lumen, the loss of B9D2 function could contribute to the abnormal bile duct development seen in these conditions.
Broader Implications and Future Research Directions
This study not only provides new insights into the mechanisms underlying biliary dysgenesis in liver-related ciliopathies but also highlights the importance of exploring the extraciliary functions of proteins involved in ciliopathies. Many ciliary proteins, including those in the transition zone, have been found to play critical roles in various cellular processes beyond the primary cilium, such as cell-cell adhesion, polarity, and signaling.
By expanding our understanding of these “moonlighting” functions of ciliary proteins, researchers may uncover additional pathways and therapeutic targets for the complex disorders associated with ciliopathies. Furthermore, this work emphasizes the need for a more comprehensive approach to studying ciliopathies, considering both the ciliary and non-ciliary roles of the proteins involved.
As the scientific community continues to delve deeper into the mysteries of ciliopathies, studies like this one will undoubtedly pave the way for a better understanding of these debilitating conditions and the development of more effective treatments.
Author credit: This article is based on research by Chloe Caenen-Braz, Latifa Bouzhir, Pascale Dupuis-Williams.
For More Related Articles Click Here