Inherited retinal diseases (IRDs) are a group of complex genetic disorders that can lead to progressive vision loss. Researchers have now uncovered a unique genetic variant in the RP1 gene that could provide important insights into the diagnosis and treatment of these debilitating conditions. The study, led by a team of scientists from South Korea, focused on identifying the prevalence and diagnostic challenges associated with a specific Alu insertion in the RP1 gene, which has been linked to a form of retinitis pigmentosa (RP).
Unraveling the Genetic Complexity of Inherited Retinal Diseases
IRDs are a diverse group of genetic disorders that affect the retina, the light-sensitive tissue at the back of the eye. These conditions can lead to a range of vision problems, from night blindness and progressive vision loss to complete blindness. Identifying the underlying genetic causes of IRDs is crucial for accurate diagnosis, genetic counseling, and the development of targeted therapies.
The Prevalence and Diagnostic Challenges of the RP1 Alu Insertion
The researchers analyzed genetic data from 1,072 individuals of Korean descent, including 411 patients with IRDs and 661 patients with other genetic disorders. They found that the Alu insertion in the RP1 gene was present in 1.5% of the IRD patients, but not in the non-IRD group. This suggests that the Alu insertion is relatively rare in the general population, but a significant contributor to IRDs, particularly a form of RP.
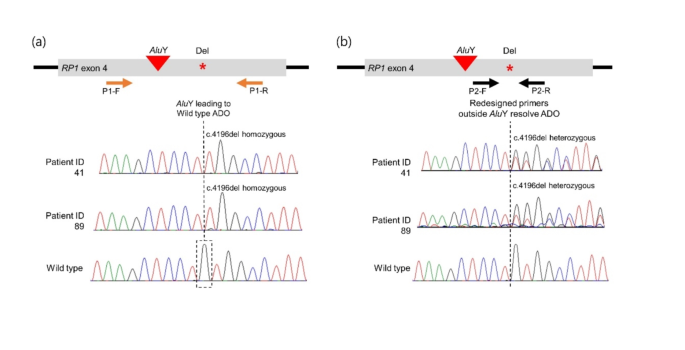
However, the researchers also identified several diagnostic challenges associated with the Alu insertion. For example, the insertion can lead to allele dropout during standard genetic testing, resulting in the incorrect identification of certain RP1 variants. The researchers developed an allele-specific PCR technique to accurately detect the Alu insertion and its relationship with other RP1 variants.
Implications for Genetic Diagnosis and Therapeutic Strategies
The study’s findings have important implications for the genetic diagnosis of IRDs. By understanding the prevalence and diagnostic challenges of the RP1 Alu insertion, clinicians can implement more accurate and efficient testing strategies to identify the underlying genetic causes of these complex disorders.
Furthermore, the discovery of this founder mutation in the RP1 gene may pave the way for the development of targeted therapies for a subset of RP patients. As the researchers note, “Delineating diagnostic challenges of AluY insertion and strategies to avoid potential pitfalls could aid clinicians in an accurate molecular diagnosis for patients with IRD.”

Unraveling the Genetic Diversity of Retinitis Pigmentosa
Retinitis pigmentosa is the most common form of IRD, affecting an estimated 2 million people worldwide. This condition is characterized by the progressive degeneration of the rod and cone photoreceptors in the retina, leading to night blindness, tunnel vision, and eventually, complete vision loss.
The genetic basis of RP is highly complex, with mutations in more than 80 genes known to be associated with the condition. The RP1 gene, which encodes a protein essential for the stability and organization of the photoreceptor outer segments, is one of the genes commonly implicated in both autosomal dominant and autosomal recessive forms of RP.
Deciphering the Impact of the RP1 Alu Insertion
The Alu insertion identified in this study is a specific type of genetic variation known as a mobile element insertion. These insertions can disrupt the normal function of a gene and lead to genetic disorders. In the case of the RP1 Alu insertion, the researchers found that it results in a premature stop codon in the RP1 protein, likely leading to a non-functional or truncated form of the protein.
Interestingly, the researchers also found that the RP1 Alu insertion often occurs in combination with other pathogenic variants in the RP1 gene, resulting in a compound heterozygous state. This means that individuals with the Alu insertion also carry a second, distinct RP1 variant on the other allele, further complicating the genetic diagnosis and inheritance pattern of the disease.

Overcoming Diagnostic Challenges with Innovative Strategies
The study’s findings highlight the importance of understanding the impact of genetic variations, such as the RP1 Alu insertion, on the accuracy of genetic testing and diagnosis. The researchers developed a specialized allele-specific PCR technique to accurately detect the Alu insertion and its relationship with other RP1 variants, overcoming the diagnostic challenges posed by this genetic feature.
Additionally, the researchers utilized long-read sequencing technology to determine the phase of different RP1 variants, providing crucial information about the allelic status of the mutations. This is particularly important for accurately diagnosing autosomal recessive forms of RP, where the pathogenic variants must be present on separate alleles.
Towards Personalized Genetic Diagnosis and Targeted Therapies
The findings from this study underscore the need for a comprehensive and personalized approach to the genetic diagnosis of IRDs. By understanding the prevalence and impact of specific genetic variations, such as the RP1 Alu insertion, clinicians can develop more accurate and efficient testing strategies to identify the underlying genetic causes of these complex disorders.
Moreover, the discovery of this founder mutation in the RP1 gene may pave the way for the development of targeted therapies for a subset of RP patients. As the researchers note, “Delineating diagnostic challenges of AluY insertion and strategies to avoid potential pitfalls could aid clinicians in an accurate molecular diagnosis for patients with IRD.”
As the field of genomic medicine continues to evolve, studies like this one will be crucial in unraveling the genetic complexities of inherited retinal diseases and ultimately improving the lives of those affected by these debilitating conditions.
Author credit: This article is based on research by Mi-Ae Jang, Jong Kwon Lee, Jong-Ho Park, Sungsoon Hwang, Young-gon Kim, Jong-Won Kim, Youn-Ji Hong, Sang Jin Kim, Ja-Hyun Jang.
For More Related Articles Click Here